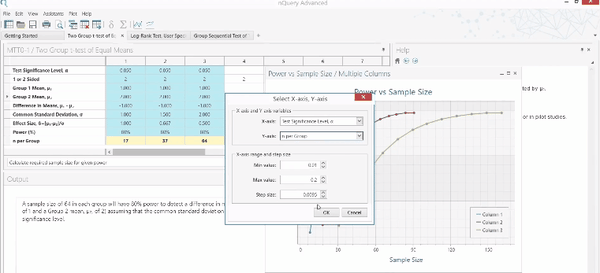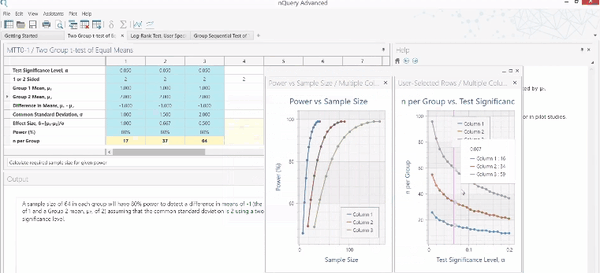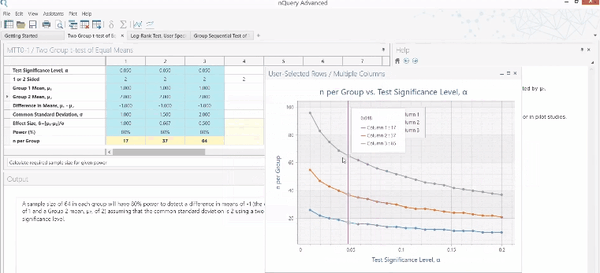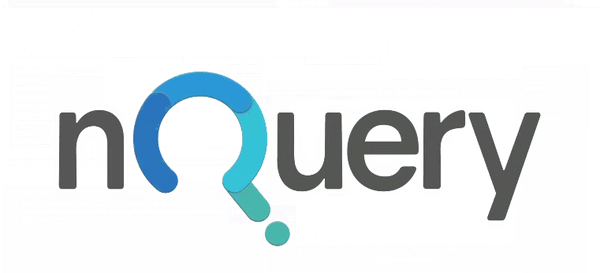
The calculation of the correct sample size is one of the first and most important steps in study design. Below is a list of sample size determination practices to be avoided as per the E9 Statistical Principles for Clinical Trials found in the FDA Guidance for industry.

The calculation of the correct sample size is one of the first and most important steps in study design. Below is a list of sample size determination practices to be avoided as per the E9 Statistical Principles for Clinical Trials found in the FDA Guidance for industry.
Before we continue, let us recap what is required for sample size estimation.
Using the usual method for determining the appropriate sample size, the following items need to be specified:
- A primary variable(s)
- The test statistic(s)
- The null hypothesis
- The alternative (working) hypothesis at the chosen dose(s) i.e.
- Effect size
- The probability of erroneously rejecting the null hypothesis (The type I error)
- The probability of erroneously failing to reject the null hypothesis (The type II error)
- Adjustments for treatment withdrawals and protocol violations
How many subjects do I need to obtain
a significant result for my study?
The most common question posed to a biostatistician
Mistake #1
Failing To Fully Examine Existing Research
Novice researchers often do not spend the appropriate amount of time examining existing literature that may of already addressed part of the question they wish to research.
Important insights can be gained from these before proceeding with your trial. You will most likely have many internal resources available to search a large number of databases and journals.
In addition to paid resources, there are many available to the public such as:
Mistake #2
Failing To Design Techniques That Avoid Bias
The most important design techniques for avoiding bias in clinical trials are blinding and randomization. These should be normal features of most controlled clinical trials.
Blinding
It is desirable for trials to follow a double-blind approach. A double-blind trial is one in which neither the subject nor any of the investigator or sponsor staff involved in the treatment or clinical evaluation of the subjects are aware of the treatment received.
Treatments are prepacked in accordance with a suitable randomization schedule, and supplied to the trial center(s) labelled only with the subject number and the treatment period, so that no one involved in the conduct of the trial is aware of the specific treatment allocated to any particular subject, not even as a code letter.
If a double-blind trial is not feasible, then the single-blind option should be considered. In some cases only an open-label trial is practically or ethically possible. In some cases only an open-label trial is practically or ethically possible.
Bias can also be reduced at the design stage by specifying procedures in the protocol aimed at minimizing any anticipated irregularities in trial conduct that might impair a satisfactory analysis, including various types of protocol violations, withdrawals and missing values. The protocol should consider ways both to reduce the frequency of such problems and to handle the problems that do occur in the analysis of data.
Randomization
Randomization introduces a deliberate element of chance into the assignment of treatments to subjects in a clinical trial. In combination with blinding, randomization helps to avoid possible bias in the selection and allocation of subjects arising from the predictability of treatment assignments.
Although unrestricted randomization is an acceptable approach, some advantages can generally be gained by randomizing subjects in blocks.
This helps to increase the comparability of the treatment groups, particularly when subject characteristics may change over time, as a result, for example, of changes in recruitment policy.
Mistake #3
Not Using Multiple Primary Variables Where Necessary
It may sometimes be desirable to use more than one primary variable, each of which (or a subset of which) could be sufficient to cover the range of effects of the therapies. The planned manner of interpretation of this type of evidence should be carefully spelled out.
It should be clear whether an impact on any of the variables, some minimum number of them,or all of them, would be considered necessary to achieve the trial objectives.
The primary hypothesis or hypotheses and parameters of interest (e.g. mean, percentage,distribution) should be clearly stated with respect to the primary variables identified, and the approach to statistical inference described. The effect on the Type I error should be explained because of the potential for multiplicity problems and the method of controlling Type I error should be given in the protocol.
The extent of intercorrelation among the proposed primary variables may be considered in evaluating the impact on Type I error. If the purpose of the trial is to demonstrate effects on all of the designated primary variables, then there is no need for adjustment of the Type I error, but the impact on Type II error and sample size should be carefully considered.
Mistake #4
Not Accounting For The Loss Of Power
From Categorized Variables
Dichotomization or other categorization of continuous or ordinal variables may sometimes be desirable. Criteria of success and response are common examples of dichotomies that should be specified precisely in terms of, for example, a minimum percentage improvement (relative to baseline) in a continuous variable or a ranking categorized as at or above some threshold level (e.g., good) on an ordinal rating scale.
The reduction of diastolic blood pressure below 90 mmHg is a common dichotomization. Categorizations are most useful when they have clear clinical relevance.
The criteria for categorization should be predefined and specified in the protocol, as knowledge of trial results could easily bias the choice of such criteria.
As categorization normally implies a loss of information, a consequence will be a loss of power in the analysis; this should be accounted for in the sample size calculation.
Mistake #5
Changes In Inclusion And Exclusion Criteria
Inclusion and exclusion criteria should remain constant, as specified in the protocol, throughout the period of subject recruitment.
Changes may occasionally be appropriate, for example, in long-term trials, where growing medical knowledge either from outside the trial or from interim analyses may suggest a change of entry criteria.
Changes may also result from the discovery by monitoring staff that regular violations of the entry criteria are occurring or that seriously low recruitment rates are due to over-restrictive criteria.
Changes should be made without breaking the blind and should always be described by a protocol amendment. This amendment should cover any statistical consequences, such as sample size adjustments arising from different event rates, or modifications to the planned analysis, such as stratifying the analysis according to modified inclusion/exclusion criteria.
Mistake #6
Not Preparing For Sample Size Adjustment
In long-term trials there will usually be an opportunity to check the assumptions which underlie the original design and sample size calculations. This may be particularly important if the trial specifications have been made on preliminary and/or uncertain information.
An interim check conducted on the blinded data may reveal that overall response variances, event rates or survival experience are not as anticipated.
A revised sample size may then be calculated using suitably modified assumptions, and should be justified and documented in a protocol amendment and in the clinical study report. The steps taken to preserve blindness and the consequences, if any, for the Type I error and the width of confidence intervals should be explained. The potential need for re-estimation of the sample size should be envisaged in the protocol whenever possible.
For example, a trial sized on the basis of safety questions or requirements or important secondary objectives may need larger numbers of subjects than a trial sized on the basis of the primary efficacy question.
Mistake #7
Incorrectly Reporting Sample Size
According to the CONSORT statement, sample size calculations should be reported and justified in all published Randomised Control Trials (RCTs). Correct output statements should be applied in many situations in addition to RCTs, such as funding applications.
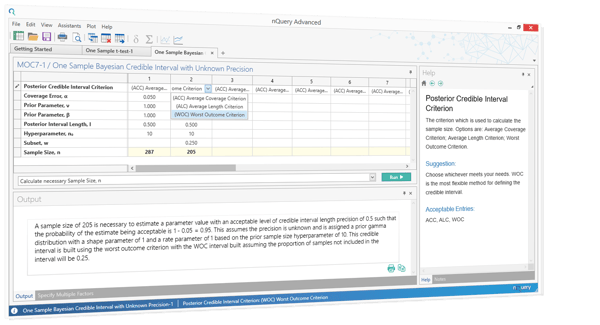
Output statements are almost meaningless if they neglect full details such as the estimates for the effect of interest and the variability. nQuery will automatically write up your sample size statement in the correct language and the format required for regulatory agencies.
Sources: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm073137.pdf
About nQuery Sample Size Software:
nQuery is now the world's most trusted sample size and power analysis software.
In 2017, 90% of organizations with FDA approved clinical trials used nQuery as their sample size calculator. It is used by Biostatisticians of all levels of expertise. Created by sample size experts, nQuery boasts an extensive list of easy-to-use but powerful feature for sample size calculation and power analysis.